|
|||||||||||||||
Все документы, представленные в каталоге, не являются их официальным изданием и предназначены исключительно для ознакомительных целей. Электронные копии этих документов могут распространяться без всяких ограничений. Вы можете размещать информацию с этого сайта на любом другом сайте.
ГОСТ 30404-2000
(ИСО 157-96)
|
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ |
ТОПЛИВО ТВЕРДОЕ МИНЕРАЛЬНОЕ
Определение форм серы
МЕЖГОСУДАРСТВЕННЫЙ СОВЕТ
ПО СТАНДАРТИЗАЦИИ, МЕТРОЛОГИИ И СЕРТИФИКАЦИИ
Минск
Предисловие
1. РАЗРАБОТАН Институтом горючих ископаемых, Технический Комитет по стандартизации ТК 179 «Твердое минеральное топливо»
ВНЕСЕН Госстандартом России
2. ПРИНЯТ Межгосударственным Советом по стандартизации, метрологии и сертификации (протокол № 17-2000 от 22.06.2000 г.)
За принятие проголосовали:
|
Наименование государства |
Наименование национального органа по стандартизации |
|
Азербайджанская Республика |
Азгосстандарт |
|
Республика Армения |
Армгосстандарт |
|
Республика Беларусь |
Госстандарт Беларуси |
|
Республика Казахстан |
Госстандарт Республики Казахстан |
|
Киргизская Республика |
Киргизстандарт |
|
Российская Федерация |
Госстандарт России |
|
Республика Таджикистан |
Таджикгосстандарт |
|
Туркменистан |
Главная государственная инспекция Туркменистана |
|
Республика Узбекистан |
Узгосстандарт |
|
Украина |
Госстандарт Украины |
3. Настоящий стандарт представляет собой аутентичный текст международного стандарта ИСО 157-1996 «Уголь. Определение форм серы» и содержит дополнительные требования, отражающие потребности экономики страны
4. Постановлением Государственного комитета Российской Федерации по стандартизации и метрологии от 2 октября 2000 г. № 243-ст межгосударственный стандарт ГОСТ 30404-2000 введен в действие непосредственно в качестве государственного стандарта Российской Федерации с 1 июля 2001 г.
5. ВЗАМЕН ГОСТ 30404-94
Содержание
Введение
Во многих случаях бывает достаточно определить массовую долю общей серы в твердом топливе, но иногда необходимо установить, как сера распределяется между органической и минеральной массой топлива.
Общая сера - сумма разных видов серы в органической и минеральной массах топлива.
Сера в угле обычно встречается в трех формах:
а) сульфатная - часть общей серы, входящая в состав неорганических сульфатов;
б) пиритная - часть общей серы, входящая в состав пиритов и марказита;
в) органическая - часть общей серы, входящая в состав органических соединений.
|
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ |
ТОПЛИВО ТВЕРДОЕ МИНЕРАЛЬНОЕОпределение форм серы.Solid
mineral fuels. |
Дата введения 2001-07-01
1 Общие требования
1.1. Область применения
Настоящий стандарт распространяется на бурые и каменные угли, антрациты, лигниты, торф, кокс и горючие сланцы (далее - топливо) и устанавливает методы определения массовых долей сульфатной, пиритной и органической серы.
Дополнительные требования, отражающие потребности экономики страны, выделены курсивом.
1.2. Нормативные ссылки
В настоящем стандарте использованы ссылки на следующие стандарты:
ГОСТ 199-78 Натрий уксуснокислый 3-водный. Технические условия
ГОСТ 1277-75 Серебро азотнокислое. Технические условия
ГОСТ 1770-74 Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Технические условия
ГОСТ 2059-95 (ИСО 351-84) Топливо твердое минеральное. Метод определения общей серы сжиганием при высокой температуре
ГОСТ 3118-77 Кислота соляная. Технические условия
ГОСТ 3760-79 Аммиак водный. Технические условия
ГОСТ 4108-72 Барий хлорид 2-водный. Технические условия
ГОСТ 4145-74 Калий сернокислый. Технические условия
ГОСТ 4204- 77 Кислота серная. Технические условия
ГОСТ 4220-75 Калий двухромовокислый. Технические условия
ГОСТ 4461-77 Кислота азотная. Технические условия
ГОСТ 4478-78 Кислота сульфосалициловая 2-водная. Технические условия
ГОСТ 4919.1-77 Реактивы и особо чистые вещества. Методы приготовления растворов индикаторов
ГОСТ 5456-79 Гидроксиламина гидрохлорид. Технические условия
ГОСТ 6613-86 Сетки проволочные тканые с квадратными ячейками. Технические условия
ГОСТ 8606-93 (ИСО 334-92) Топливо твердое минеральное. Определение общей серы. Метод Эшка
ГОСТ 9147-80 Посуда и оборудование лабораторные фарфоровые. Технические условия
ГОСТ 10398-76 Реактивы и особо чистые вещества. Комплексонометрический метод определения содержания основного вещества
ГОСТ 10652-73 Соль динатриевая этилендиамин - N, N, N`, N`-тетрауксусной кислоты 2-водная (трилон Б)
ГОСТ 10678-76 Кислота ортофосфорная термическая. Технические условия
ГОСТ 10742-71 Угли бурые, каменные, антрацит, горючие сланцы и угольные брикеты. Методы отбора и подготовки проб для лабораторных испытаний
ГОСТ 10929-76 Водорода пероксид. Технические условия
ГОСТ 11303-75 Торф и продукты его переработки. Метод приготовления аналитических проб
ГОСТ 11305-83 Торф. Методы определения влаги
ГОСТ 12026-76 Бумага фильтровальная лабораторная. Технические условия
ГОСТ 23083-78 Кокс каменноугольный, пековый и термоантрацит. Методы отбора и подготовки проб для испытаний
ГОСТ 25336-82 Посуда и оборудование лабораторные стеклянные. Типы, основные параметры и размеры
ГОСТ 27313-95 (ИСО 1170-77) Топливо твердое минеральное. Обозначения показателей качества и формулы пересчета результатов анализа для различных состояний топлива
ГОСТ 27314-91 (ИСО 589-81) Топливо твердое минеральное. Методы определения влаги
ГОСТ 27589-91 (ИСО 687-74) Кокс. Метод определения влаги в аналитической пробе
ГОСТ 29169-91 Посуда лабораторная стеклянная. Пипетки с одной отметкой
ГОСТ 29252-91 Посуда лабораторная стеклянная. Бюретки без времени ожидания
1.3 Сущность методов
Методы определения форм серы основаны на различной растворимости сульфатов и пиритов в растворах соляной и азотной кислот в определенных условиях. Сущность методов заключается в последовательном переведении сульфатов и пиритов в раствор и их прямом определении.
Общая схема анализа приведена на рисунке 1.
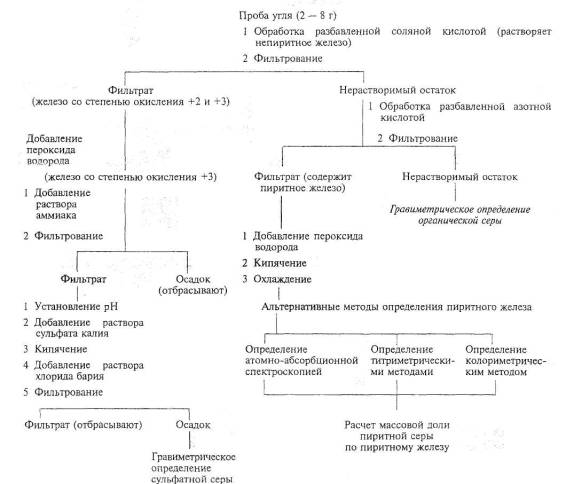
Примечание: Сера органическая = Сера общая - (Сера сульфатная + Сера пиритная)
Рисунок 1 - Общая схема анализа
1.4. Подготовка пробы
Для анализа используют аналитическую пробу, приготовленную по ГОСТ 10742, ГОСТ 23083 или ГОСТ 11303 в зависимости от вида топлива.
25 г аналитической пробы измельчают до прохождения через сито с размером отверстий приблизительно 75 мкм (сита 008 или 0071 по ГОСТ 6613). Пробу подсушивают до воздушно-сухого состояния. Для этого разложенную тонким слоем пробу выдерживают на воздухе в течение минимального времени, необходимого для достижения равновесия между влажностью пробы и окружающей атмосферы.
Перед взятием навески для определения серы воздушно-сухую пробу тщательно перемешивают не менее 1 мин предпочтительно механическим способом.
Одновременно берут отдельные навески для определения общей серы по ГОСТ 8606 и влаги по ГОСТ 27314, ГОСТ 27589 или ГОСТ 11305 в зависимости от вида топлива.
2 Разделение сульфатной и пиритной серы
2.1. Сущность метода
Сульфатную серу и непиритное железо растворяют в разбавленной соляной кислоте. Пиритную серу и пиритное железо, остающиеся в нерастворимом остатке, отделяют фильтрованием.
2.2. Реактивы
Все реактивы должны быть квалификации ч.д.а. Для анализа применяют только дистиллированную воду.
2.2.1. Соляная кислота по ГОСТ 3118, концентрированная, плотностью 1,18 г/см3, 36 %-ный раствор (по массе).
2.2.2. Соляная кислота, 15 %-ный раствор (по массе)
Разбавляют 420 см3 соляной кислоты (2.2.1) водой до 1 дм3.
2.2.3. Азотная кислота по ГОСТ 4461, 9 %-ный раствор (по массе)
Разбавляют 130 см3 концентрированной азотной кислоты (70 %-ный раствор) водой до 1 дм3.
2.3. Аппаратура
Используют стеклянную и фарфоровую посуду по ГОСТ 1770, ГОСТ 9147 и ГОСТ 25336.
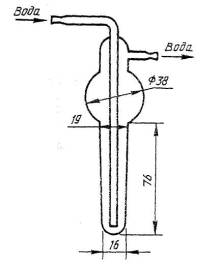
Рисунок 2 – Пальчиковый холодильник
2.3.1. Весы аналитические, точность взвешивания до 0,1 мг. Допускается применять аналитические весы с точностью взвешивания до 0,2 мг.
2.3.2. Пальчиковый холодильник, свободно входящий в горловину конической колбы вместимостью 250 см3 (рисунок 2).
2.3.3. Обратный холодильник типа ХШ-1 или 3 по ГОСТ 25336.
2.4 Проведение испытания
2.4.1. Навеска
Масса навески указана в таблице 1 в зависимости от ожидаемой массовой доли общей серы. Навеску взвешивают с точностью до 0,1 мг.
Таблица 1
|
Массовая доля общей серы, % |
Масса навески, г |
|
До 0,7 От 0,7 » 2,0 включ. Св. 2,0 |
8 5 2 |
2.4.2. Экстракция
Навеску пробы помещают в коническую колбу вместимостью 250 см3, добавляют 50 см3 раствора соляной кислоты (2.2.2.) и вставляют в горловину колбы пальчиковый (2.3.2) или обратный (2.3.3.) холодильник. Допускается проводить экстракцию в химическом стакане без холодильника; в этом случае объем раствора соляной кислоты увеличивают до 100 см3. Суспензию кипятят в течение 30 мин, поддерживая при этом постоянный ток воды в холодильнике. Снимают холодильник, тщательно обмывают его водой, собирая промывные воды в эту же коническую колбу.
Суспензию фильтруют в высокий стакан вместимостью 400 см3 через фильтр средней плотности, предварительно дважды промытый кислотой. Остаток на фильтре промывают три раза раствором соляной кислоты (2.2.2.), а затем три раза горячей дистиллированной водой порциями приблизительно по 30 см3. В фильтрате определяют сульфатную серу. Фильтр с остатком переносят в коническую колбу вместимостью 250 см3, добавляют 50 см3 раствора азотной кислоты (2.2.3.) и оставляют для определения пиритной серы.
3 Определение сульфатной серы
3.1. Сущность метода
Сульфатную серу, экстрагированную из навески пробы раствором соляной кислоты, определяют гравиметрически, осаждая хлоридом бария.
3.2. Реактивы
Все реактивы должны быть квалификации ч.д.а. Для анализа применяют только дистиллированную воду.
3.2.1. Пероксид водорода по ГОСТ 10929, 30 %-ный раствор (по массе).
3.2.2. Метиловый красный по ГОСТ 4919.1, индикатор, раствор
Растворяют 1 г натриевой соли 2-(4-диметиламинофенилазо)-бензойной кислоты в 1 дм3 воды.
3.2.3. Гидроксид аммония по ГОСТ 3760, концентрированный раствор не менее 25 % (по массе), плотностью приблизительно 0,8 г/см3.
3.2.4. Соляная кислота по 2.2.1.
3.2.5. Сульфат калия по ГОСТ 4145, раствор 2 г/дм3
Растворяют 2 г сульфата калия в воде и разбавляют до 1 дм3.
3.2.6. Хлорид бария по ГОСТ 4108, раствор 85 г/дм3
Растворяют 100 г двуводного хлорида бария в воде и разбавляют водой до 1 дм3. Перед использованием раствор фильтруют через плотный фильтр, дважды промытый кислотой.
3.2.7. Нитрат серебра по ГОСТ 1277, раствор 17 г/дм3
Растворяют 17 г нитрата серебра в воде, добавляют несколько капель концентрированной азотной кислоты и разбавляют водой до 1 дм3.
3.3. Аппаратура
Используют стеклянную и фарфоровую посуду по ГОСТ 1770, ГОСТ 9147 и ГОСТ 25336.
3.3.1. Весы аналитические (2.3.1.).
3.3.2. Печь муфельная электрическая с терморегулятором для поддержания температуры (800±25)°С и воздухообменом.
3.3.3. Тигли из платины, кварца или глазурованного фарфора вместимостью приблизительно 25 см3.
3.3.4. Пластина толщиной 6 мм из кварца или огнеупорного материала, которая легко вставляется в муфельную печь (3.3.2).
3.3.5. Конус для фильтрования фарфоровый, диаметром 25 мм.
3.4. Проведение испытания
К фильтрату, полученному в результате экстракции навески раствором соляной кислоты (2.4.2.), добавляют 5 см3 раствора пероксида водорода (3.2.1.) и кипятят в течение 5 мин для превращения всего растворенного железа в трехвалентное. Для осаждения железа в горячий раствор добавляют 2-3 капли раствора индикатора метилового красного (3.2.2.), затем по каплям приливают раствор гидроксида аммония (3.2.3.) до щелочной реакции раствора (желтый цвет) плюс 5 капель в избыток. Образовавшуюся суспензию фильтруют через неплотный фильтр в стакан вместимостью 400 см3. Осадок гидроксида железа тщательно промывают горячей водой и отбрасывают. К фильтрату осторожно по каплям добавляют концентрированную соляную кислоту (3.2.4.) до перехода окраски раствора в розовую и затем приливают еще 1 см3 кислоты. Общий объем раствора должен составлять 150-250 см3.
С помощью пипетки с одной меткой по ГОСТ 29169 к фильтрату приливают 25,0 см3 раствора сульфата калия (3.2.5.). Стакан накрывают часовым стеклом и нагревают до закипания содержимого стакана, затем снижают температуру нагрева до прекращения кипения. В середину горячего раствора при перемешивании приливают в течение 20 с пипеткой 10 см3 холодного раствора хлорида бария (3.2.6.). В течение последующих 30 мин раствор выдерживают при температуре, близкой к температуре кипения, без перемешивания.
Раствор фильтруют одним из перечисленных ниже способов.
а) Фильтрование под действием силы тяжести через беззольный плотный фильтр по ГОСТ 12026 диаметром 100-125 мм, дважды промытый кислотой. Фильтр помещают в химическую воронку типа 3-75-110 (170) по ГОСТ 25336. Стебель воронки so время фильтрования должен быть заполнен жидкостью.
б) Фильтрование под действием силы тяжести через фильтровальную прокладку, приготовленную из беззольной дважды промытой кислотой фильтровальной бумаги.
Для приготовления фильтровальной прокладки дважды промытую кислотой фильтровальную бумагу в виде кусочков размером около 1 см2 встряхивают в сосуде с водой до полного ее разрушения. Помещают конус для фильтрования (3.3.5.) в фильтровальную воронку диаметром 75 мм, закрывают стебель воронки пальцем и приливают воду так, чтобы она покрыла фильтровальный конус и заполнила стебель воронки. Помещают бумажную массу в фильтровальный конус, встряхиванием воронки добиваются, чтобы масса образовала прокладку толщиной 5 мм, поверхность прокладки разравнивают стеклянной палочкой с расплющенным концом. Убирают палец и дают избытку воды стечь, затем фильтровальную прокладку слегка утрамбовывают по краям и отжимают с помощью стеклянной палочки. Фильтр еще раз промывают водой, после чего он готов к работе.
После переноса фильтровальной прокладки в тигель воронку дважды протирают кусочками беззольной фильтровальной бумаги, которую затем сжигают вместе с фильтровальной прокладкой.
Осадок на фильтре промывают горячей водой до полного удаления ионов хлора, т.е. пока порция промывных вод объемом 20 см3 даст лишь незначительную опалесценцию при добавлении раствора нитрата серебра (3.2.7.). Общий объем промывных вод должен составлять не более 250 см3.
Влажный фильтр или фильтровальную прокладку переносят в предварительно прокаленный и взвешенный тигель (3.3.3.) и слегка уплотняют. Тигель помещают на холодную пластину (3.3.4.), вставляют в муфельную печь (3.3.2.) и прокаливают при температуре (800±25)°С в течение 15 мин. Чтобы предотвратить унос при озолении, влажный фильтр предварительно высушивают и обугливают над горелкой или на электрической плитке, не допуская воспламенения. В этом случае прокаливать тигель с осадком можно без пластины. После озоления тигель вынимают из муфельной печи, охлаждают сначала на воздухе, а затем в эксикаторе и взвешивают.
3.5. Контрольный опыт
Проводят контрольный опыт, как указано в 2.4.2. и 3.4., но без навески пробы.
3.6. Обработка результатов
Массовую
долю сульфатной серы в пробе ![]() %, вычисляют по формуле
%, вычисляют по формуле
![]()
где m1 - масса навески, взятой для экстракции соляной кислотой, г;
m2 - масса сульфата бария, полученная при определении, г;
m3 - масса сульфата бария, полученная в контрольном опыте, г.
Примечание: Расчет коэффициента, используемого в данной формуле, приведен в приложении А.
За результат испытаний принимают среднее арифметическое результатов двух определений с точностью до 0,01 %.
3.7. Точность
3.7.1. Сходимость
Результаты двух определений, проведенных в разное время в одной лаборатории одним исполнителем с использованием одной и той же аппаратуры из представительных навесок одной и той же аналитической пробы, не должны отличаться более чем на 0,02 абс. %. Результаты должны быть пересчитаны на одинаковую массовую долю влаги.
3.7.2. Воспроизводимость
Средние результаты параллельных определений, проведенных в двух разных лабораториях из представительных порций, отобранных из одной и той же пробы после последней стадии ее приготовления, не должны отличаться более чем на 0,03 абс. % при уровне доверительной вероятности 95 %. Результаты должны быть пересчитаны на одинаковую массовую долю влаги.
3.7.3. Если расхождение между результатами двух определений превышает указанные значения, то проводят третье определение и за результат принимают среднее арифметическое результатов двух наиболее близких результатов в пределах допускаемых расхождений.
Если результат третьего определения находится в пределах допускаемых расхождений по отношению к каждому из двух предыдущих определений, то за результат анализа принимают среднее арифметическое результатов трех определений.
4 Определение пиритной серы
4.1. Сущность метода
Остаток, нерастворимый в соляной кислоте (2.4.2.), экстрагируют разбавленной азотной кислотой для перевода в раствор пиритного железа. В экстракте железо определяют одним из прямых методов: титриметрическим, колориметрическим или методом атомно-абсорбционной спектрометрии. Массовую долю пиритной серы в исходной пробе рассчитывают по массовой доле пиритного железа, учитывая стехиометрическое соотношение 1:2 в химической формуле пирита FeS2.
Экстракцию пиритного железа азотной кислотой проводят одним из альтернативных методов: при кипячении в течение 30 мин, при комнатной температуре в течение 24 ч или при комнатной температуре в течение 2 ч при непрерывном встряхивании.
Остаток, нерастворимый в соляной и азотной кислотах, при необходимости может быть использован для определения органической серы.
Примечание: Этот метод может быть использован для определения пиритной серы в углях всех типов. Однако при кипячении бурых углей, лигнитов и торфов с азотной кислотой получаются окрашенные экстракты. Если окраска экстракта не исчезает при кипячении с пероксидом водорода, то титриметрическое определение железа в экстракте становится невозможным. В этом случае для определения железа используют колориметрический метод или метод атомно-абсорбционной спектрометрии, или обработку топлив азотной кислотой проводят при комнатной температуре.
4.2. Реактивы
Все реактивы должны быть квалификации ч.д.а. Для анализа применяют только дистиллированную воду.
4.2.1. Для титриметрического, колориметрического методов и метода атомно-абсорбционной спектрометрии
4.2.1.1. Азотная кислота по 2.2.3.
4.2.1.2. Пероксид водорода по 3.2.1.
4.2.2. Только для титриметрического метода
4.2.2.1. Гидроксид аммония по 3.2.3.
4.2.2.2. Соляная кислота по 2.2.2.
4.2.2.3. Хлорид олова (II), раствор 50 г/дм3.
Растворяют 5 г безводного хлорида олова (III) или 6 г двуводной соли в 50 см3 концентрированной соляной кислоты (2.2.1.) при нагревании. Раствор вливают в 40 см3 воды, охлаждают и разбавляют водой до 100 см3.
Раствор готовят непосредственно перед использованием.
4.2.2.4. Хлорид ртути (II), насыщенный раствор 7 г хлорида ртути (II) вносят в 100 см3 воды и перемешивают смесь в течение 10 мин.
4.2.2.5. Смесь серной и ортофосфорной кислот
Осторожно приливают 150 см3 серной кислоты по ГОСТ 4204 (раствор 98 %) к 500 см3 воды. После охлаждения смеси приливают 150 см3 раствора ортофосфорной кислоты по ГОСТ 10678 (раствор 85 %) и разбавляют раствор до 1 дм3 водой.
4.2.2.6. Дифениламиносульфонат натрия по ГОСТ 4919.1, индикатор, раствор 2 г/дм3 Растворяют 0,2 г дифениламиносульфоната натрия в воде и разбавляют до 100 см3.
Раствор хранят в посуде из темного стекла.
4.2.2.7. Калий двухромовокислый (бихромат) по ГОСТ 4220, стандартный раствор для титрования, с (K2Сr2O7) = 0,003 моль/дм3.
Растворяют 0,8826 г бихромата калия (предварительно высушенного в течение 2 ч при 150°С) в воде в мерной колбе вместимостью 1 дм3, а затем доливают водой до метки.
4.2.2.8. Метиловый красный по 3.2.2.
4.2.2.9. Кислота сульфосалициловая (C7H6O6S·2H2O) no ГОСТ 4478, раствор 10 %-ный (по массе).
4.2.2.10. Соль динатриевая этилендиамин-N,N,N'N'-тетрауксусной кислоты 2-водная (трилон Б), по ГОСТ 10652, раствор 0,05 моль/дм3.
Приготовление раствора из стандарт-титра или по ГОСТ 10398
4.2.3. Для методов колориметрии и атомно-абсорбционной спектрометрии
4.2.3.1. Железо, исходный стандартный раствор
Растворяют 0,1000 г чистого железа в виде проволоки в 2,5 см3 концентрированной азотной кислоты (70 %-ный раствор) и 7,5 см3 воды. Кипятят до удаления оксидов азота, охлаждают и разбавляют водой до метки в мерной колбе вместимостью 100 см3. 1 см3 исходного стандартного раствора содержит 1 мг железа.
4.2.4. Только для колориметрического метода
4.2.4.1. Хлорид гидроксиламина по ГОСТ 5456, раствор 100 г/дм3 Растворяют 10 г хлорида гидроксиламина в воде и разбавляют до 100 см3.
4.2.4.2. Конго красный по ГОСТ 4919.1, индикаторная бумага.
4.2.4.3. Ацетат натрия по ГОСТ 199, раствор 328 г/дм3
Растворяют 32,8 г безводного ацетата натрия в воде и разбавляют до 100 см3.
4.2.4.4. Фенантролин по ГОСТ 4919.1, индикатор, раствор
Растворяют 0,625 г гидрата или солянокислого 1,10-фенантролина в воде и разбавляют до 250 см3.
Раствор хранят в посуде из темного стекла и выбрасывают в случае появления бурой окраски.
4.2.5. Только для метода атомно-абсорбционной спектрометрии
4.2.5.1. Хлорид лантана (III), раствор 100 г/дм3
Растворяют 267 г 7-водного хлорида лантана (III) в воде и разбавляют до 1 дм3.
4.3. Аппаратура
Используют стеклянную и фарфоровую посуду по ГОСТ 1770, ГОСТ 9147, ГОСТ 25336 и ГОСТ 29252.
4.3.1. Весы аналитические по 2.3.1.
4.3.2. Пальчиковый холодильник по 2.3.2.
4.3.3. Обратный холодильник по 2.3.3.
4.3.4. Спектрофотометр (для работы в ультрафиолетовой и видимой области спектра) для измерения оптической плотности при длине волны 510 нм.
4.3.5. Атомно-абсорбционный спектрометр для измерения абсорбции при длинах волн 248,3 и 372,0 нм.
4.4. Проведение испытания
4.4.1. Приготовление раствора для анализа
В конической колбе вместимостью 250 см3, в которой ранее проводили экстракцию соляной кислотой (см. 2.4.2.), остаток и фильтр, помещенные в азотную кислоту, перемешивают с помощью стеклянной палочки с расплющенным концом, измельчая при этом фильтр. Затем конец палочки обмывают и палочку удаляют. В соответствии с примечанием к 4.1. остаток в растворе азотной кислоты оставляют стоять при комнатной температуре на 24 ч или перемешивают на встряхивателе в течение 2 ч, или в горловину колбы вставляют пальчиковый (4.3.2.) или обратный (4.3.3.) холодильник и кипятят содержимое колбы в течение 30 мин при постоянном токе воды в холодильнике. Снимают холодильник, тщательно обмывают его, собирая промывные воды в коническую колбу. Содержимое колбы фильтруют в стакан через фильтр средней плотности, дважды промытый кислотой. Остаток на фильтре промывают три раза раствором азотной кислоты (4.2.1.1.), а затем три раза горячей водой, при этом каждая порция промывного раствора или воды составляет около 30 см3. Фильтрат и промывные воды собирают в один стакан. Нерастворимый остаток при необходимости может быть использован для определения органической серы.
Добавляют к фильтрату 5 см3 раствора пероксида водорода (4.2.1.2.) и кипятят в течение 5 мин для обесцвечивания фильтрата. Затем фильтрат охлаждают и определяют в нем железо (III) одним из методов, приведенных в 4.4.2., 4.4.3. или 4.4.4.
4.4.2. Титриметрический метод
4.4.2.1. Подготовка раствора для титрования
Нагревают раствор по 4.4.1 до кипения и добавляют 2-3 капли раствора индикатора метилового красного (4.2.2.8.). Для осаждения гидроксида железа (III) к кипящему раствору медленно, по каплям, приливают раствор гидроксида аммония (4.2.2.1.) до небольшого избытка (желтое окрашивание), а потом еще 5 капель в избыток. Смесь фильтруют через неплотный фильтр. Осадок гидроксида железа тщательно промывают горячей водой с несколькими каплями аммиака. Для количественного перенесения осадка гидроксида железа (III) фильтр прокалывают и смывают осадок струей горячей воды в колбу или стакан вместимостью 500 см3. Для удаления с фильтра следов гидроксида железа промывают фильтр небольшими порциями горячей разбавленной соляной кислоты (4.2.2.2.), а затем горячей воды. Для полного растворения гидроксида железа содержимое колбы нагревают. Далее железо (III) определяют в растворе по 4.4.2.2. или 4.4.2.3.
4.4.2.2. Бихроматное титрование железа в растворе
Раствор, полученный при растворении гидроксида железа (III) в соляной кислоте, нагревают и выпаривают до объема 20 см3. В горячий раствор при перемешивании добавляют по каплям помощью пипетки или капельницы раствор хлорида олова (II) (4.2.2.3.) до исчезновения желтого окрашивания. Добавляют еще 5 капель в избыток, раствор быстро охлаждают до комнатной температуры и быстро приливают 10 см3 раствора хлорида ртути (II) (4.2.2.4.). Образуется мелки осадок хлорида ртути (I).
Добавляют 15 см3 смеси серной и ортофосфорной кислот (4.2.2.5.), разбавляют раствор водой до объема 150-200 см3, добавляют 5 капель раствора индикатора дифениламиносульфоната натри (4.2.2.6.) и титруют стандартным раствором бихромата калия (4.2.2.7.). Около конечной точки титрования цвет раствора становится голубовато-зеленым, а в присутствии большого количеств железа - зеленовато-голубым. На этой стадии титрования раствор бихромата калия добавляют по каплям до перехода окраски раствора в не изменяющийся интенсивный фиолетово-голубой цвет. Отмечают объем израсходованного титранта в см3.
Примечание: При анализе проб, содержащих более 2 % (по массе) пиритной серы, для титрования необходимы большие количества стандартного раствора бихромата калия. В этих случаях раствор для титрования (4.4.2.1.) переносят в мерную колбу, разбавляют водой до метки и отбирают пипеткой аликвоту. Разбавление учитывают при расчете.
4.4.2.3. Комплексонометрическое титрование железа в растворе
Раствор, полученный при растворении гидроксида железа (III) в соляной кислоте, нейтрализуют раствором гидроксида аммония (4.2.2.1.), приливая его по каплям до появления осадка. Затем приливают при перемешивании по каплям раствор соляной кислоты (4.2.2.2.) до рН 1,4-1,8 и добавляют 1-2 см3 раствора сульфосалициловой кислоты (4.2.2.9.). Раствор нагревают до 55-60ºС и в горячем состоянии, титруют раствором трилона Б (4.2.2.10.) до перехода красно-фиолетовой окраски в зеленовато-желтую или до обесцвечивания раствора в зависимости от массовой доли железа.
Допускается комплексонометрическое титрование железа без предварительного выделения гидроксида железа в бесцветном фильтрате, полученном по 4.4.1.
4.4.3. Колориметрический метод
4.4.3.1. Приготовление калибровочных растворов
С помощью пипетки с одной меткой переносят 20 см3 стандартного раствора железа (4.2.3.1.) в мерную колбу вместимостью 500 см3, разбавляют водой до метки и тщательно перемешивают. Переносят аликвоты полученного раствора 1, 2, 3 ... 10 см3 (отобранные с помощью пипеток с одной меткой) в отдельные мерные колбы вместимостью 50 см3. Разбавляют каждую аликвоту до 25 см3 водой, добавляют по 5 см3 раствора хлорида гидроксиламина (4.2.4.1.) и по небольшом; кусочку индикаторной бумаги конго красный (4.2.4.2.). Смесь титруют раствором ацетата натрия (4.2.4.3.), пока индикаторная бумага не станет красной. Добавляют 4 см3 раствора фенантролина (4.2.4.4.) и разбавляют до 50 см3 водой. Смесь тщательно перемешивают и оставляют на 1 ч.
4.4.3.2. Определение
Раствор, подготовленный для определения пиритного железа (см. 4.4.1.), переносят в мерную колбу вместимостью 250 см3, разбавляют водой до метки и тщательно перемешивают.
С помощью пипетки аликвоту α см3 (см. таблицу 2) полученного раствора переносят в мерную колбу вместимостью 50 см3 раствора хлорида гидроксиламина (4.2.4.1.) и небольшой кусочек индикаторной бумаги конго красный (4.2.4.2.). Смесь титруют раствором ацетата натрия (4.2.4.3.) до тех пор, пока индикаторная бумага не станет красной. Добавляют 4 см3 раствора индикатора фенантролина (4.2.4.4.) и разбавляют водой до 50 см3. Смесь тщательно перемешивают и оставляют на 1 ч.
Таблица 2
|
Предполагаемая массовая доля пиритной серы, % |
Аликвота а для колориметрического определения железа, см3 |
|
До 0,7 От 0,7 и более |
2 1 |
Измеряют оптическую плотность калибровочных растворов (4.4.3.1.) и полученного раствора с помощью спектрофотометра (4.3.4.) при длине волны 510 нм в кюветах подходящего размере относительно воды. По результатам анализа калибровочных растворов (4.4.3.1) строят калибровочный график в координатах: оптическая плотность, измеренная на спектрофотометре - масса железа в калибровочных растворах (в мкг). С помощью калибровочного графика по оптической плотности разбавленного испытуемого раствора находят соответствующее количество железа (в мкг).
4.4.4. Метод атомно-абсорбционной спектрометрии.
4.4.4.1. Приготовление калибровочных растворов
Готовят серию калибровочных растворов, перекрывающих ожидаемую область измеряемых концентраций. Для этого переносят соответствующие объемы стандартного раствора железа (4.2.3.1.) в отдельные мерные колбы вместимостью 100 см3. В каждую колбу добавляют 20 см3 разбавленной азотной кислоты (4.2.1.1.) и 4 см3 раствора хлорида лантана (III) (4.2.5.1.). Разбавляют водой до метки и тщательно перемешивают.
4.4.4.2. Определение
К приготовленному для анализа раствору (см. 4.4.1.) добавляют 10 см3 раствора хлорида лантана (III) (4.2.5.1.). Раствор разбавляют водой до метки в мерной колбе вместимостью 250 см3 и тщательно перемешивают.
Атомно-абсорбционный спектрометр настраивают на длину волны, соответствующую железу. После необходимой регулировки горелки в соответствии с инструкцией по эксплуатации прибора зажигают пламя.
Параметры, которые необходимо соблюдать при определении железа, приведены в таблице 3.
Таблица 3
|
Параметр |
Значение для области измерений, мг Fe/дм3 |
|
|
0-100 |
0-9 |
|
|
Длина волны, нм Ширина щели, нм Длина горелки, мм Пламя |
372,0 0,2 50 Воздух/ацетилен* |
248,3 0,2 50 Воздух/ацетилен * |
|
* Соотношение в соответствии с инструкцией по эксплуатации прибора. |
||
Измеряют абсорбцию калибровочных растворов (4.4.4.1.) и раствора, получение которого описано в данном пункте, с помощью атомно-абсорбционного спектрометра (4.3.5.), регистрируя показания прибора после стабилизации величины абсорбции. Измерения проводят не менее двух раз. По результатам анализа калибровочных растворов (4.4.4.1.) строят калибровочный график в координатах: абсорбция - концентрация железа в калибровочных растворах (в мкг/см3). С помощью калибровочного графика по значению абсорбции находят концентрацию железа в разбавленном испытуемом растворе (в мкг/см3).
Примечания:
1. Для приведения показания спектрометра к нулю используют воду. После проведения каждого измерения горелку промывают водой.
2. Если концентрация железа в анализируемом растворе превышает область измерений, исходный раствор следует дополнительно разбавить. При этом концентрации хлорида лантана и азотной кислоты в разбавленных растворах должны соответствовать концентрациям в исходном растворе.
4.5. Контрольный опыт
Одновременно готовят раствор для контрольного опыта по 2.4.2. и 4.4.1., но без навески пробы. Определяют концентрацию железа в контрольном растворе по 4.4.2., 4.4.3. или 4.4.4.
4.6. Обработка результатов
4.6.1. Титриметрический метод (см. 4.4.2.).
4.6.1.1. При определении пиритной серы с
использованием бихроматного титрования (см. 4.4.2.2.) массовую долю пиритной серы в аналитической
пробе ![]() , %, вычисляют по формуле
, %, вычисляют по формуле
![]() (2)
(2)
где V1 - объем раствора бихромата калия, израсходованного на титрование пробы, см3;
V2 - объем раствора бихромата калия, израсходованного на титрование контрольного раствора, см3;
m1 - масса исходной навески пробы, взятой для экстракции соляной кислотой (см. 2.4.1.), г.
Примечание: Расчет коэффициента приведен в приложении А
4.6.1.2. При определении пиритной серы с
использованием комплексонометрического титрования (см. 4.4.2.3.) массовую долю пиритной серы в аналитической
пробе ![]() %, вычисляют по формуле
%, вычисляют по формуле
![]() (3)
(3)
где V3 - объем раствора ди-Na-ЭДТА, израсходованного на титрование пробы, см3;
V4 - объем раствора ди-Na-ЭДТА, израсходованного на титрование контрольного раствора, см3;
M1 - концентрация раствора ди-Na-ЭДТА, моль/дм3;
m1 - масса исходной навески пробы, взятой для экстракции соляной кислотой (см. 2.4.1.), г.
Примечание: Расчет коэффициента приведен в приложении А.
4.6.2. Колориметрический метод
При
определении пиритной серы с использованием колориметрического метода (см.
4.4.3.) массовую долю пиритной серы в аналитической пробе ![]() , %,
вычисляют по формуле
, %,
вычисляют по формуле
![]() (4)
(4)
где m4 - масса железа в разбавленном испытуемом растворе (см. 4.4.3.2), мкг;
m5 - масса железа в контрольном растворе (см. 4.5), мкг;
m1 - масса исходной навески пробы, взятой для экстракции соляной кислотой (см. 2.4.1.), г;
V5 - объем аликвоты, взятой для определения из разбавленного испытуемого раствора (см. 4.4.3.2.), см3.
Примечание: Расчет коэффициента приведен в приложении А.
4.6.3. Метод атомно-абсорбционной спектрометрии
При
определении пиритной серы методом атомно-абсорбционной спектрометрии (см. 4.4.4.) массовую долю пиритной серы в
аналитической пробе ![]() , %, вычисляют
по формуле
, %, вычисляют
по формуле
![]() (5)
(5)
где ρFe,1 - концентрация железа в разбавленном испытуемом растворе (см. 4.4.4.2.), мкг/см3;
ρFe,1 - концентрация железа в контрольном растворе (см. 4.5.), мкг/см3;
m1 - масса исходной навески пробы, взятой для экстракции соляной кислотой (см. 2.4.1.), г.
Примечание: Расчет коэффициента приведен в приложении А.
4.6.4 Результат определения, равный среднему арифметическому результатов двух параллельных определений, записывают с точностью до 0,01 %.
4.7 Точность
4.7.1 Сходимость
Расхождения между результатами двух определений, проведенных в разное время в одной лаборатории одним исполнителем с использованием одной и той же аппаратуры из представительных навесок одной и той же аналитической пробы, не должны превышать значений, указанных в таблице 4.
Таблицa 4
|
Массовая доля пиритной серы, % |
Максимально допустимые расхождения между результатами (пересчитанными на одинаковую массовую долю влаги) |
|
|
Сходимость |
Воспроизводимость |
|
|
До 0,5 От 0.5 до 1,5 Св. 1,5 |
0,05 абс. % 0,07 абс. % 5 отн. % |
0,10 абс. % 0,15 абс. % 10 отн. % |
4.7.2. Воспроизводимость
Расхождения между средними значениями результатов параллельных определений, проведенных в двух разных лабораториях из представительных порций, отобранных из одной и той же пробы после последней стадии ее приготовления, не должны превышать значений, указанных в таблице 4 при уровне доверительной вероятности 95 %.
4.7.3. Если расхождение между результатами двух определений превышает указанные значения, то проводят третье определение и за результат принимают среднее арифметическое результатов двух наиболее близких результатов в пределах допускаемых расхождений.
Если результат третьего определения находится в пределах допускаемых расхождений по отношению к каждому из двух предыдущих определений, то за результат анализа принимают среднее арифметическое результатов трех определений.
5. Определение органической серы
5.1. Сущность метода
По условиям определения, регламентированным настоящим стандартом, органическая сера не растворяется в разбавленных соляной и азотной кислотах и ее массовую долю определяют по разности между массовой долей общей серы и суммой массовых долей сульфатной и пиритной серы. При необходимости массовую долю органической серы определяют экспериментально в остатке после экстракции кислотами (4.4.1.) со смесью Эшка.
5.2. Расчет
Массовую долю органической серы в пробе в процентах рассчитывают по разности массовой доли общей серы в процентах, определенной по ГОСТ 8606 или ГОСТ 2059, и суммы массовых долей сульфатной и пиритной серы в процентах.
5.3. Определение органической серы со смесью Эшка
Фильтр с остатком после экстракции кислотами (4.4.1.) подсушивают, складывают, режут ножницами в тигель, в который предварительно помещают 3 г смеси Эшка. Содержимое тигля перемешивают и равномерно засыпают 1 г смеси Эшка. Далее поступают в соответствии с ГОСТ 8606 на метод определения общей серы.
5.4. Результат, равный среднему арифметическому результатов двух параллельных определений, записывают с точностью до 0,01 %.
6. Пересчет результатов испытаний на другие состояния топлива и протокол испытаний
6.1. Пересчет результатов на другие состояния топлива
Результаты определений относятся к воздушно-сухому топливу. Пересчет результатов на другие состояния топлива производят в соответствии с ГОСТ 27313.
6.2. Протокол испытаний
Протокол испытаний должен содержать следующие данные:
а) идентификацию пробы;
б) применяемый метод со ссылкой на настоящий стандарт;
в) дату испытаний;
г) результаты испытаний и способ их выражения;
д) особенности, замеченные при проведении анализа;
е) операции, не предусмотренные настоящим стандартом или необязательные.
ПРИЛОЖЕНИЕ А
(справочное)
Расчет коэффициентов, использованных в формулах
В настоящем приложении относительные атомные массы и относительные молекулярные массы представлены в виде химических формул и символов, заключенных в квадратные скобки. При расчетах использованы относительные атомные массы, приведенные в таблице А.1.
Таблица А.1.
|
Элемент |
Символ |
Относительная атомная масса |
|
Барий Хром Железо Кислород Калий Сера |
Ba Cr Fe О K S |
137,33 51,996 55,847 15,999 39,098 32,064 |
А.1. Сульфатная сера (3.6.)
Массовую
долю сульфатной серы в аналитической пробе ![]() , %, вычисляют по формуле
, %, вычисляют по формуле
![]() (А.1.)
(А.1.)
где m1 - масса навески, взятой для экстракции соляной кислотой, г;
m2 - масса сульфата бария, полученная при определении, г;
m3 - масса сульфата бария, полученная в контрольном опыте, г.
Следовательно,
![]() (А.2.)
(А.2.)
А.2. Пиритная сера, титриметрический бихроматный метод (4.4.2.2. и 4.6.1.1.)
Массовую
долю пиритной серы в аналитической пробе ![]() , %, вычисляют по формуле
, %, вычисляют по формуле
![]() , (А.3.)
, (А.3.)
где V1 - объем раствора бихромата калия, израсходованного на титрование пробы, см3;
V2 - объем раствора бихромата калия, израсходованного на титрование контрольного раствора, см3;
m1 - масса навески, взятой для экстракции соляной кислотой, г;
с - концентрация раствора бихромата калия (4.2.2.7.), моль/дм3.
Следовательно,
![]() (А.4.)
(А.4.)
А.3. Пиритная сера, титриметрический комплексонометрический метод (4.4.2.3 и 4.6.1.2)
Массовую долю пиритной серы в
аналитической пробе ![]() , %, вычисляют по формуле
, %, вычисляют по формуле
![]() , (А.5.)
, (А.5.)
где V3 - объем раствора трилона Б, израсходованного на титрование пробы, см3;
V4 - объем раствора трилона Б, израсходованного на титрование контрольного раствора, см3;
М1 - концентрация раствора трилона Б (4.2.2.10.), моль/дм3;
m1 - масса исходной навески пробы, взятой для экстракции соляной кислотой, г.
Следовательно,
![]() (А.6.)
(А.6.)
А.4. Пиритная сера, колориметрический метод (4.4.3. и 4.6.2.)
Массовую
долю пиритной серы в аналитической пробе ![]() ,% вычисляют по формуле
,% вычисляют по формуле
![]() (А.7)
(А.7)
где m4 - масса железа в разбавленном испытуемом растворе, мкг;
m5 - масса железа в контрольном растворе, мкг;
m1 - масса исходной навески пробы, взятой для экстракции соляной кислотой, г;
V5 - объем аликвоты, взятой для определения из разбавленного испытуемого раствора, см3;
V - объем разбавленного испытуемого раствора, см3.
Следовательно,
![]() (А.8.)
(А.8.)
А.5. Пиритная сера, метод атомно-абсорбционной спектрометрии (4.4.4. и 4.6.3.)
Массовую
долю пиритной серы в аналитической пробе ![]() ,% вычисляют по формуле
,% вычисляют по формуле
![]() (А.9.)
(А.9.)
где ρFe,1 - концентрация железа в разбавленном испытуемом растворе, мкг/см3;
ρFe,2 - концентрация железа в контрольном растворе, мкг/см3;
m1 - мacca исходной навески пробы, взятой для экстракции соляной кислотой, г;
V - объем разбавленного испытуемого раствора, см3
Следовательно,
![]() (А.10.)
(А.10.)
Ключевые слова: топливо твердое; угли бурые, каменные; антрациты; лигниты; торф; кокс; горючие сланцы; общая сера; сульфатная сера; пиритная сера; органическая сера
|
|